


미국에서 허가된 인공지능 기반 의료기기(AIMD·Artificial Intelligence–Enabled Medical Device)의 상당수가 승인 직후 1년 내 리콜 사례가 많다는 연구 결과가 나왔다.
임상 검증이 없는 제품과 상장사 제품에서 리콜이 집중되는 양상도 확인됐다.
미국 존스홉킨스대와 예일대 연구진은 지난 2024년 11월까지 미국 식품의약국(FDA) 510(k) 경로로 허가된 인공지능 의료기기 950개를 대상으로 리콜 현황을 분석했다.
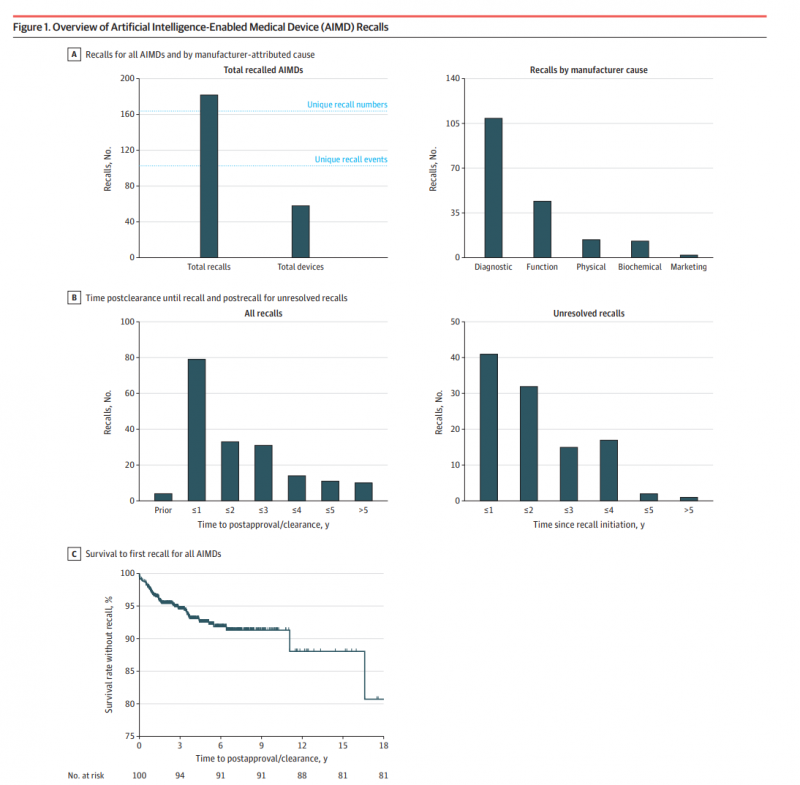
그 결과, 60개(6.3%) 기기가 총 182건 리콜에 연루됐으며, 기기당 평균 리콜 건수는 3건이었다. 리콜 원인은 진단·측정 오류가 109건으로 가장 많았고 관련 제품은 93만5063개에 달했다.
기능 지연·손실은 44건(75만5647개), 물리적 위험 14건(8192개), 생화학적 위험 13건(7만6257개)으로 집계됐다.
전체 리콜 가운데 79건(43.4%)은 승인 후 12개월 이내 발생했다. 108건(59.3%)은 조사 종료 시점까지 해결되지 않았으며, 이 중 20건은 3년 이상 미해결 상태였다.
임상 검증 여부에 따라 기기당 평균 리콜 건수는 차이를 보였다. 임상 검증이 없는 기기는 평균 3.4건이었고, 후향적 데이터 기반은 1.9건, 전향적 임상시험 기반은 2.0건이었다.
리콜 규모(평균 회수 제품 수) 또한 검증 없는 기기에서 1만2193개로 검증을 거친 기기(6523개)보다 많았다.
기업 유형별로는 상장사가 전체 인공지능 의료기기의 53.2%를 차지했지만, 리콜 91.8% 및 리콜된 제품 수의 98.7%를 차지했다.
다변량 분석 결과 임상 검증 없음(OR 2.8, 95% CI 1.6–4.7), 상장사 여부(OR 5.9, 95% CI 2.4–14.6)는 독립적으로 리콜 위험을 높이는 요인으로 나타났다.
연구진은 “현재 510(k) 절차는 인공지능 의료기기 초기 성능 문제를 충분히 걸러내지 못한다”며 전향적 임상시험 의무화, 조건부(time-limited) 허가 제도, 사후 감시(postmarket surveillance) 강화 등을 제안했다.
한편, 이번 연구는 JAMA Health Forum 8월 22일자에 게재됐다.
??
(AIMDArtificial IntelligenceEnabled Medical Device) 1 .
.
2024 11 (FDA) 510(k) 950 .
, 60(6.3%) 182 , 3. 109 935063 .
44(755647), 14(8192), 13(76257) .
79(43.4%) 12 . 108(59.3%) , 20 3 .
. 3.4, 1.9, 2.0.
( ) 12193 (6523) .
53.2% , 91.8% 98.7% .
(OR 2.8, 95% CI 1.64.7), (OR 5.9, 95% CI 2.414.6) .
510(k) , (time-limited) , (postmarket surveillance) .
, JAMA Health Forum 8 22 .









